The user can upload and analyze two types of data: either a single chromosome, or the whole cell. In this section we explain how to identify entangled structures along a single chain (a chromosome, or any other polymer-like chain with similar size): knots and slipknots.
Knot analysis can be conducted based on random, centre of mass, or direct closure methods, on two different levels:
Moreover, the user can use all these methods together with the relaxation procedure to estimate entanglement stability [1].
Entanglements of a single chromosomes are presented in the following manner. The information displayed from top to bottom of the result page can be divided into:
At the top of the page, the first tab indicates the status of a submitted job, as shown in Fig 1. The tab summarizes options used by the user to submit a job. The most important information is:
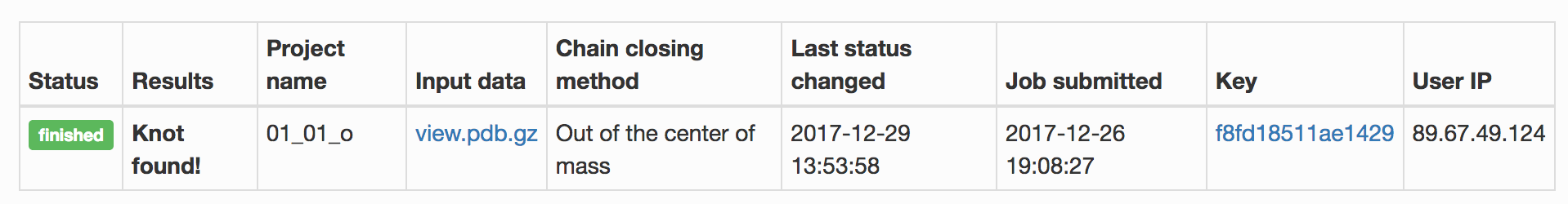

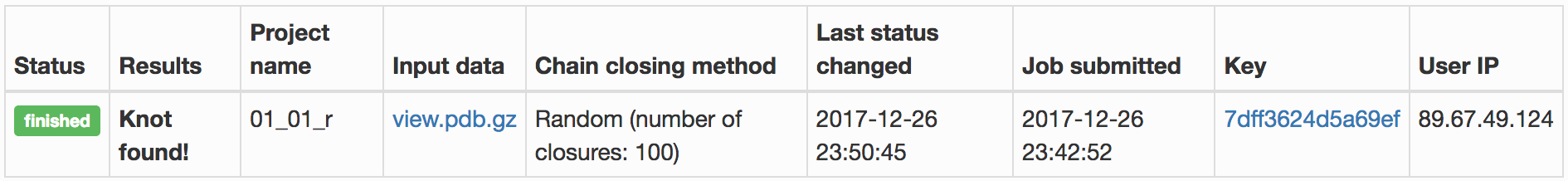

Fig. 1 Example of the job results page – interpretation of job status. Panels from top to bottom show the top of the results page for three different methods used to close the chromosome: out of the centre mass, direct, and random closure method. The last panel shows the results page for a job that is still running, when some data is already available in a form of a topological fingerprint even though the relaxation method is still running.
Users can name their jobs, hide them on the job queue, and, by supplying their e-mail addresses, request to be notified when their job finishes. E-mail addresses are stored only until the job finishes, all other data about the jobs is stored for two weeks.
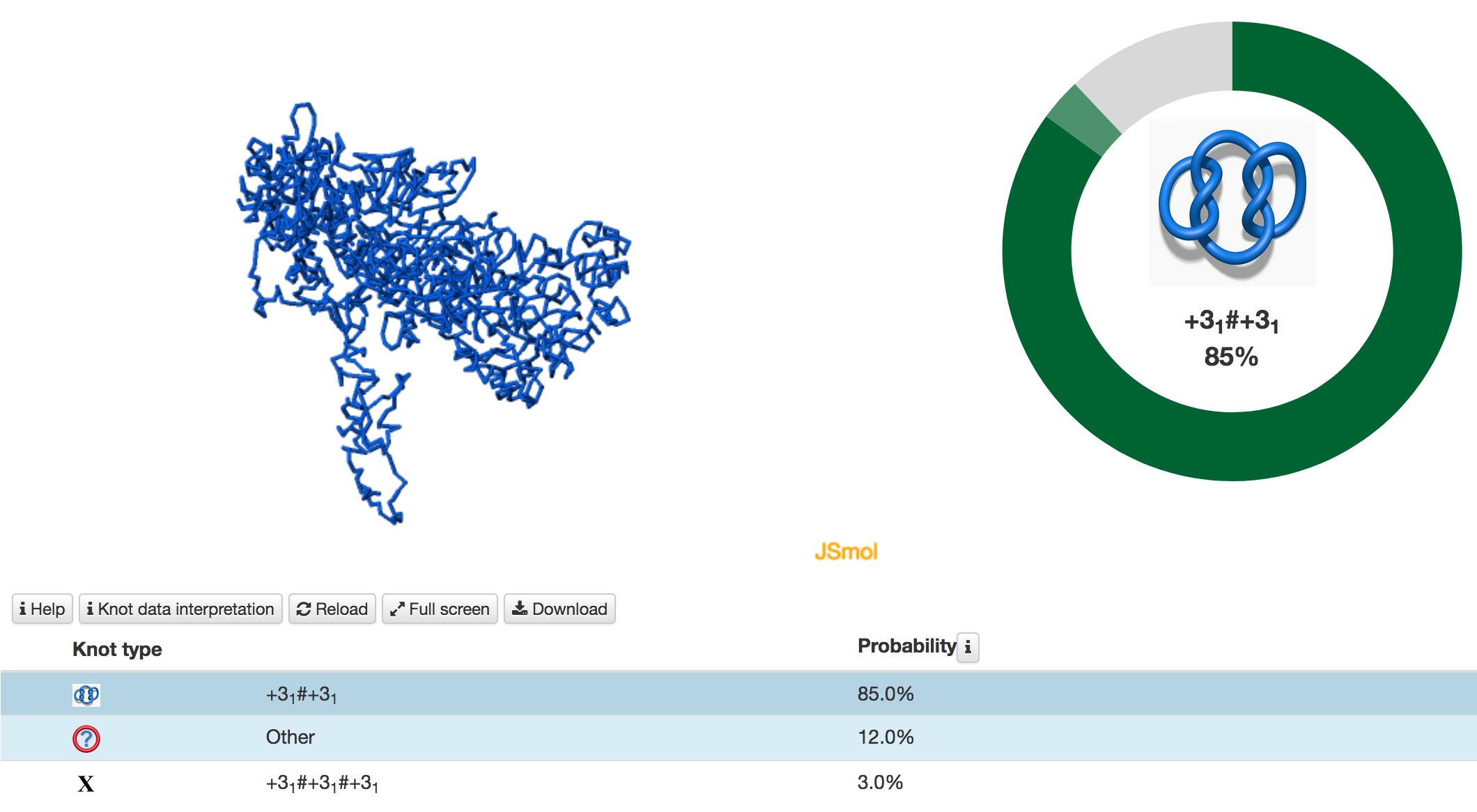
In the most advanced option critical information about the knot type and its likelihood is presented using four methods:
The main part of the default page contains the JSmol presentation of the chromosome structure and one of the two graphical representations:
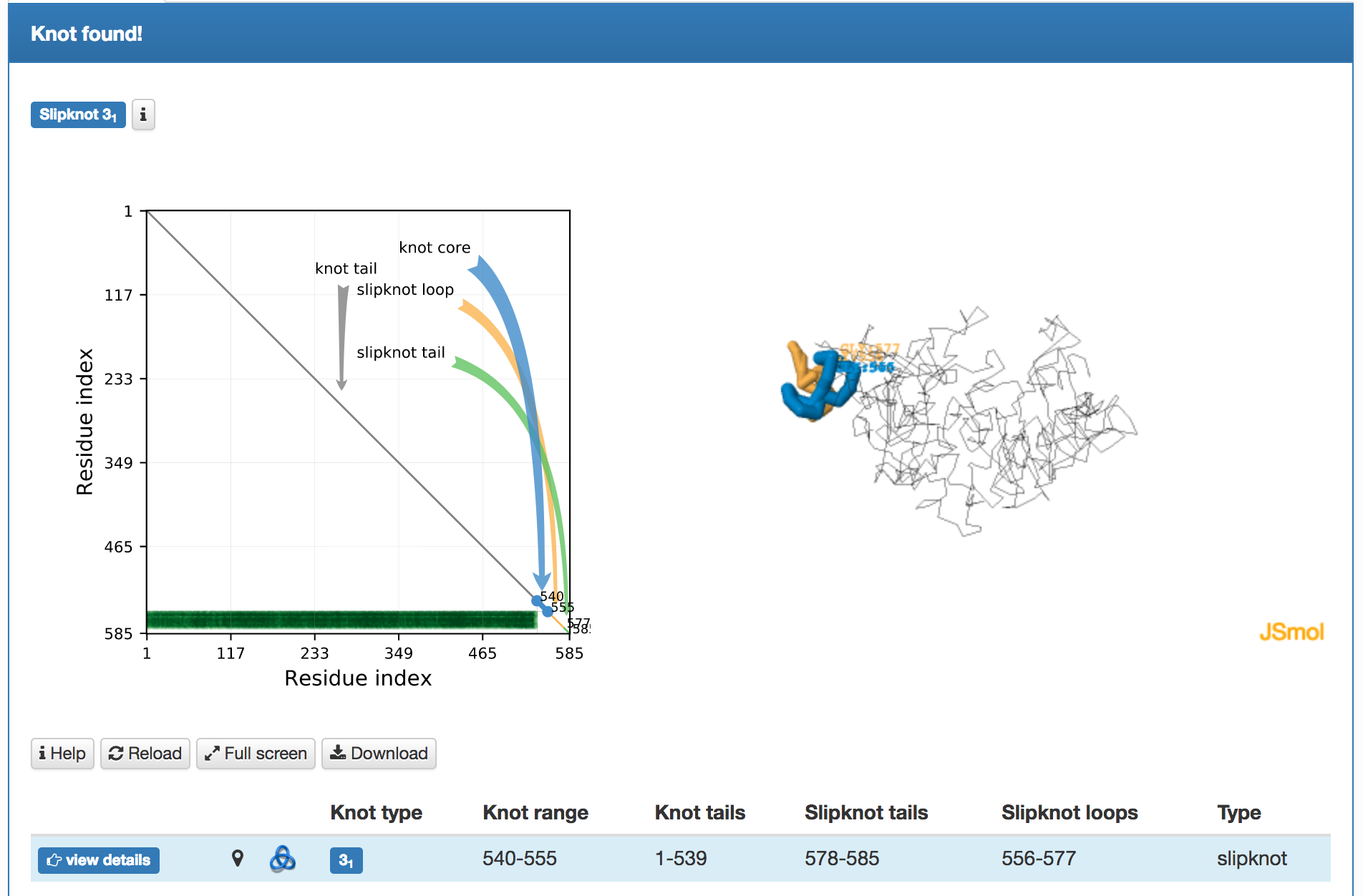
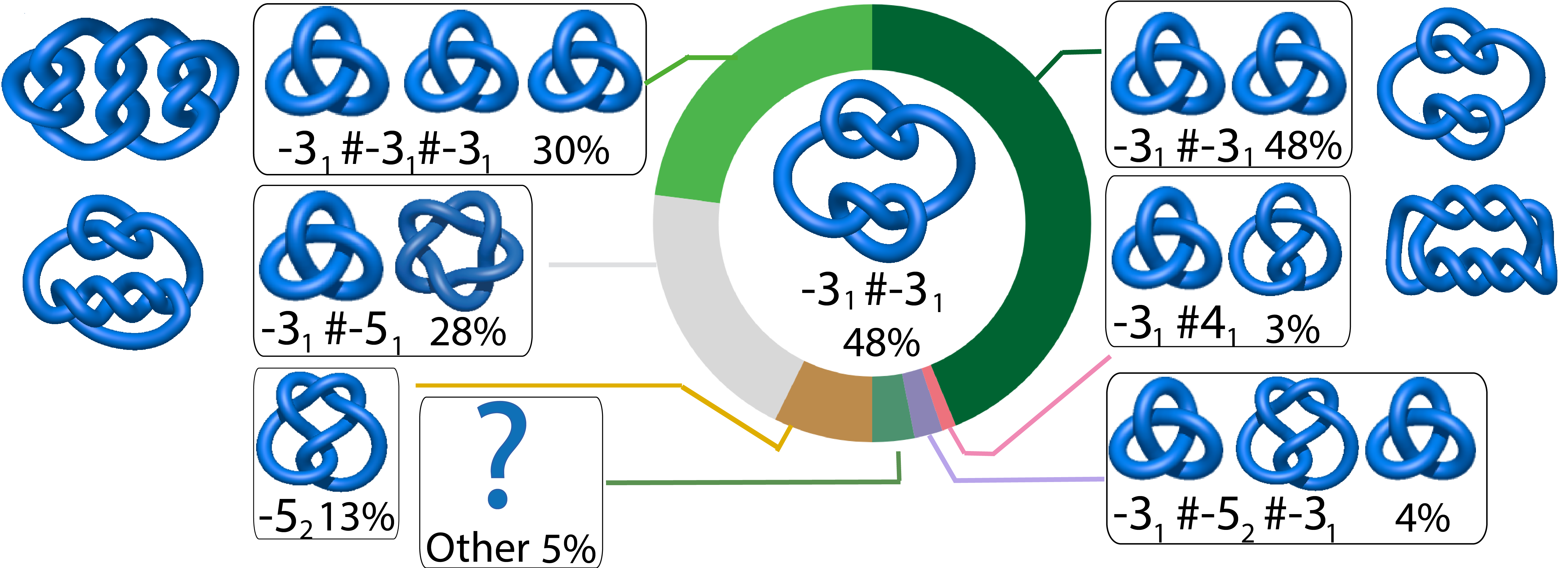
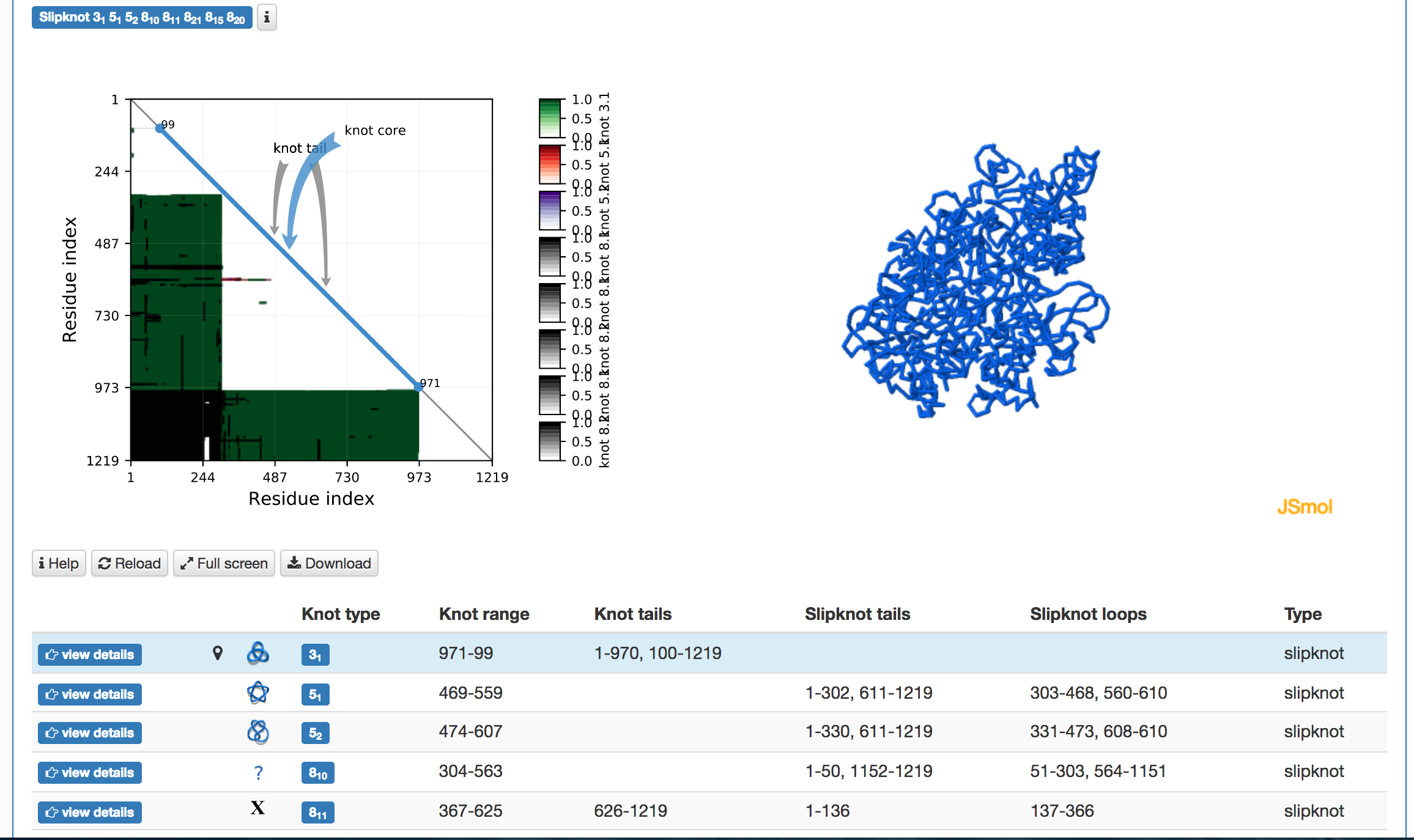
The topological and structural details about the most probable knots are stored in the table below the graphical presentation (Fig.4). Each row of the table corresponds to one knot. The rows are sorted with decreasing likelihood of the knot topology. In each row, one can find the minature depiction of the knot identified along with its name and likelihood. The rest of the table presents position of the knot core, slipknot loop, the knot tails. All this information is visible in the JSmol presentation.
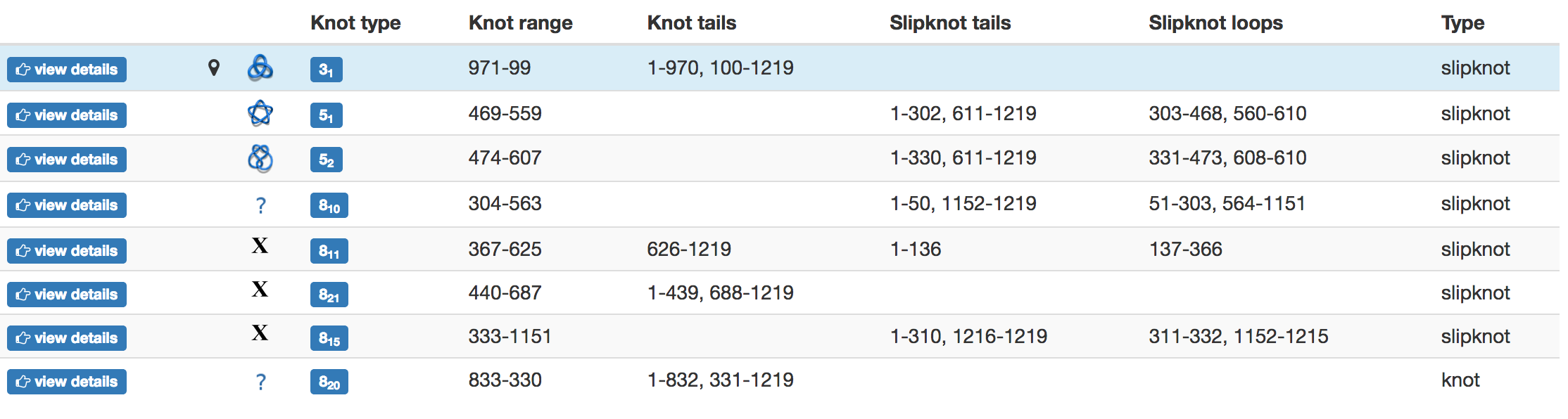
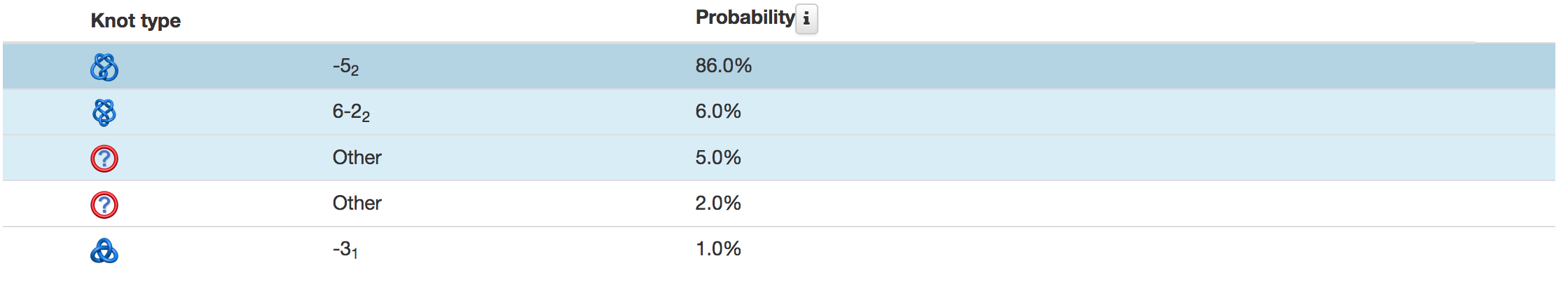
Fig. 4 Example of the table showing the observed knots in the case of the random and the direct closure method are shown in the top and bottom panel, respectively. In the case of the random closure method, the table shows detailed data about knots and slipknots type and position formed by backbone subchains. In the case of the direct closure method the table shows the knot type and probability of all identified knots.
The relaxation method is used to estimate the robustness of the topology of a given chromosome. The change in the conformation of the chromosome is investigated via molecular dynamics (it is described here) and a criterion to determine the robustness of topology is described here. The data for relaxation (Fig. 5) is presented as follows:
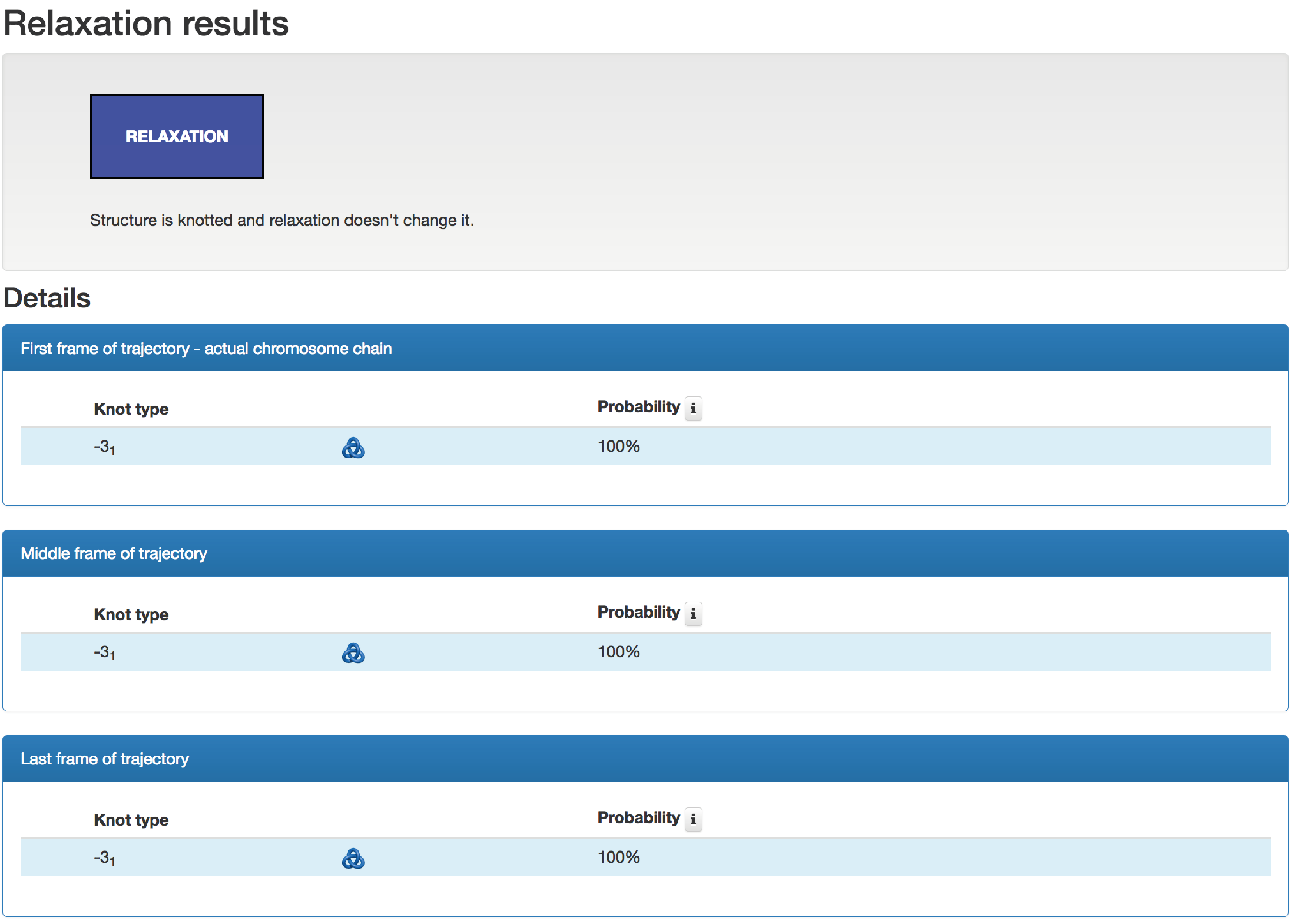
Fig. 5 Overview of the result page for a single chromosome relaxation analysis. This data shows that a sample knot remains conserved during the whole simulation.
The result of relaxation - the robustness of an investigated topology - is described via four colors and presented at the top of the page in the box called "Relaxation results":
The robustness of the topology is estimated based on the type of the topology identified in temporary conformations. The table below presents details about identified topologies in the original conformation, and middle and last frames, which were used to estimate the robustness of topology. The table contains the knot type, its schematic drawing, and probability of identified topology.