Recent developments have for the first time enabled the determination of three-dimensional structures of individual chromosomes and genomes, e.g. in nuclei of single haploid mouse embryonic [1] based on Hi-C chromosome conformation contact data. The model structures from [1] are highly knotted [2], and knots were also observed in [3]. Even though the abundance of knots in chromosomes is still controversial (as it was for a long time in the case of proteins [4,5]), there is a clear need to check model structures for entanglements.
Here we present the KnotGenom server [6] - the first server that detects and characterizes entire topology of a single chromosome, as well as all chromosomes in the cell nucleus. The KnotGenom is a powerful server, optimized to determine topology of chromosome type chains, which are 103 times longer than typical proteins. The server detects not only primary knots, but also composite knots and links. These entangled structures are determined based on computation of knot polynomials, and (in case of links) the Gaussian Linking Number (GLN). This data provides new reaction coordinates which are crucial to improve current data, to understand the geometry of chromosomes and their interactions, and to identify stable entangled configurations. In general, understanding entanglement is also a valuable resource for investigating the spatial structure-and-function relationship of genomes. The key question is what is the role of non-trivial topology (knots and links) on the level of a chromosome and what is their role in regulating gene expression.
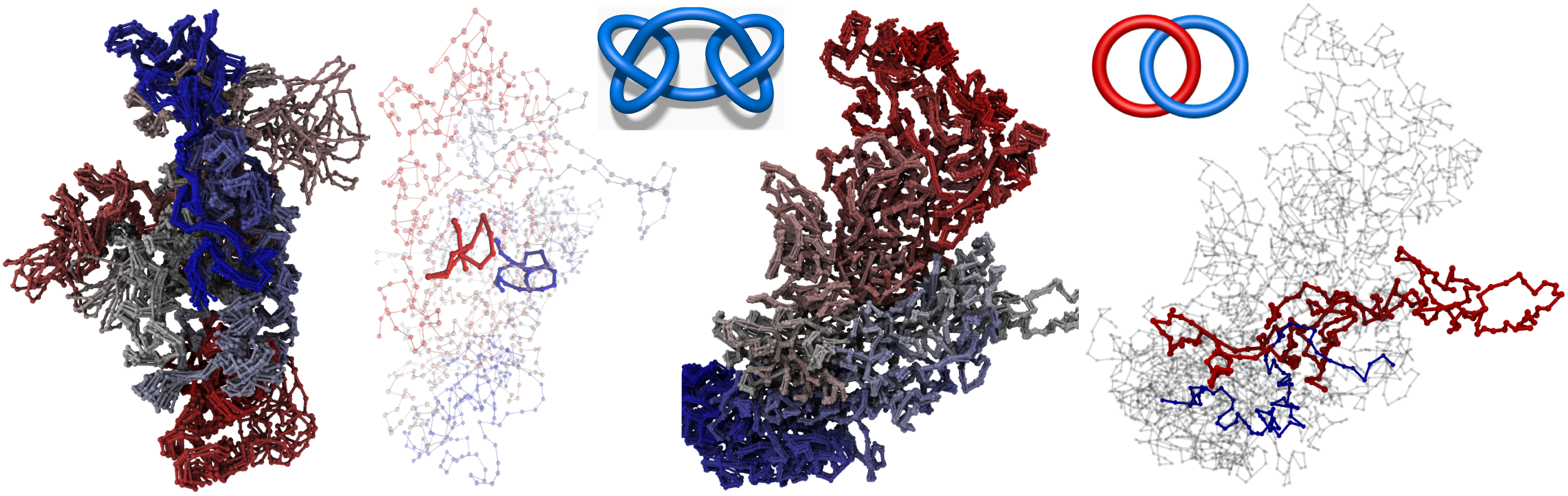
Fig. 1 Left: structures of single chromosome 14 from cell 2 [1] with an identified composite knot. Right: structure of chromosomes 5 and 9 from cell 2 [1] with an identified link.
The input data required to analyze topology of chromosome chains consists of a file with atomistic coordinates, in PDB or XYZ format (each monomer corresponding to, e.g., 100000 base pairs). The server distinguishes between a single or a pair of chromosomes, and the whole cell. A user can choose various methods to close chains, as well as the level of detail of the topological analysis ranging from simple knot determination to a complete topological fingerprint with different resolutions. The Gaussian Linking Number (GLN) method is used to spot locations of winding of chromosomes. The user can also relax model data via molecular dynamics simulations before analysis. Results are visualized via interactive matrices. Output data provides detailed information for each knot, slipknot, and link, which can be downloaded for further analysis. We also offer an option to visualize entanglements.
First, the algorithm to determine knots, knotting fingerprints, links, and GLN was validated earlier on all proteins deposited in the PDB [4,5,7]. Second, all methods that we use were developed further, optimized to take into account properties of chromosomes (especially their size), and validated on a single chromosome of length 1600 and 103 pairs (8 cells) [2]. These results, including all topological information, are also available in the job section of the KnotGenom.
We stress that the analysis provided by the KnotGenome cannot be performed by any other available servers that analyze proteins' structure and topology.